Paper by Thierry Ouisse 2018
The paper "Electronic and vibrational properties of V2C-based MXenes: From experiments to first-principles modeling" has been published in Phys. Rev. B
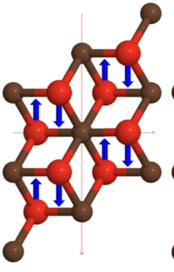
Here you will find the paper by Thierry Ouisse
"In the present work, the electronic and vibrational properties of both pristine V2C and fully terminated V2CT2 (where T=F, O, OH) two-dimensional monolayers are investigated using density functional theory. First, the atomic structures of V2C-based MXene phases are optimized, and their respective dynamical stabilities are discussed. Second, electronic band structures are computed indicating that V2C is metallic as well as all the corresponding functionalized systems. Third, the vibrational properties (phonon frequencies and spectra) of V2C-based MXenes are computed with the density functional perturbation theory and reported. Both Raman-active (Eg, A1g) and infrared-active (Eu, A2u) vibrational modes are predicted ab initio with the aim to correlate the experimental Raman peaks with the calculated vibrational modes and to assign them to specific atomic motions. The effect of the terminal groups on the vibrational properties is emphasized, along with the effect on the presence and position of the corresponding Raman peaks. Our results provide insights for the identification and characterization of V2C-based samples using Raman spectroscopy."